SeedFold Scaling Biomolecular Structure Prediction
A next-generation folding model that scales up model capacity through width scaling and large-scale data distillation. We also provide SeedFold-Linear, a more efficient variant with linear triangular attention. Both models achieve state-of-the-art results on FoldBench, outperforming AlphaFold3 on most protein-related tasks.
Three Pillars of Scaling
We scale folding models from three perspectives to achieve state-of-the-art performance
Model Scaling
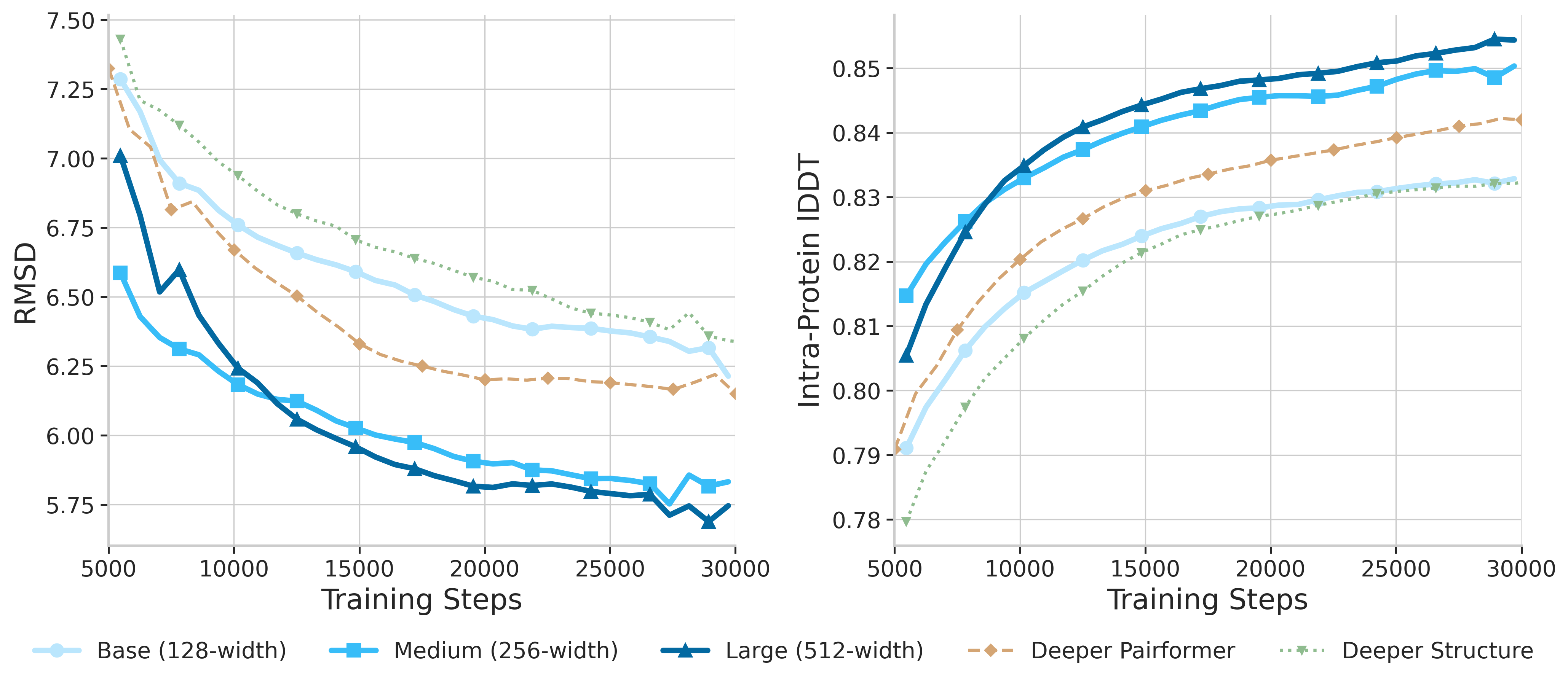
Scale the Pairformer width from 128 to 512, increasing model capacity. Training such wide networks poses significant challenges including memory constraints and training instability—we developed engineering solutions to overcome these obstacles.
- Width scaling > Depth scaling
- 128 → 256 → 384 → 512 dimensions
- Solved training stability challenges
Linear Triangular Attention
A novel attention mechanism that reduces computational complexity from O(n³) to O(n²), enabling efficient scaling while maintaining prediction quality.
- Additive & Gated variants
- 2-3× memory reduction
- Optimized Triton kernels
Large-Scale Distillation
Construct a 26.5M sample dataset through distillation from AlphaFold2, expanding training data by 147× compared to experimental structures.
- PDB + AFDB + Mgnify
- 147× data expansion
- Diverse sequence coverage
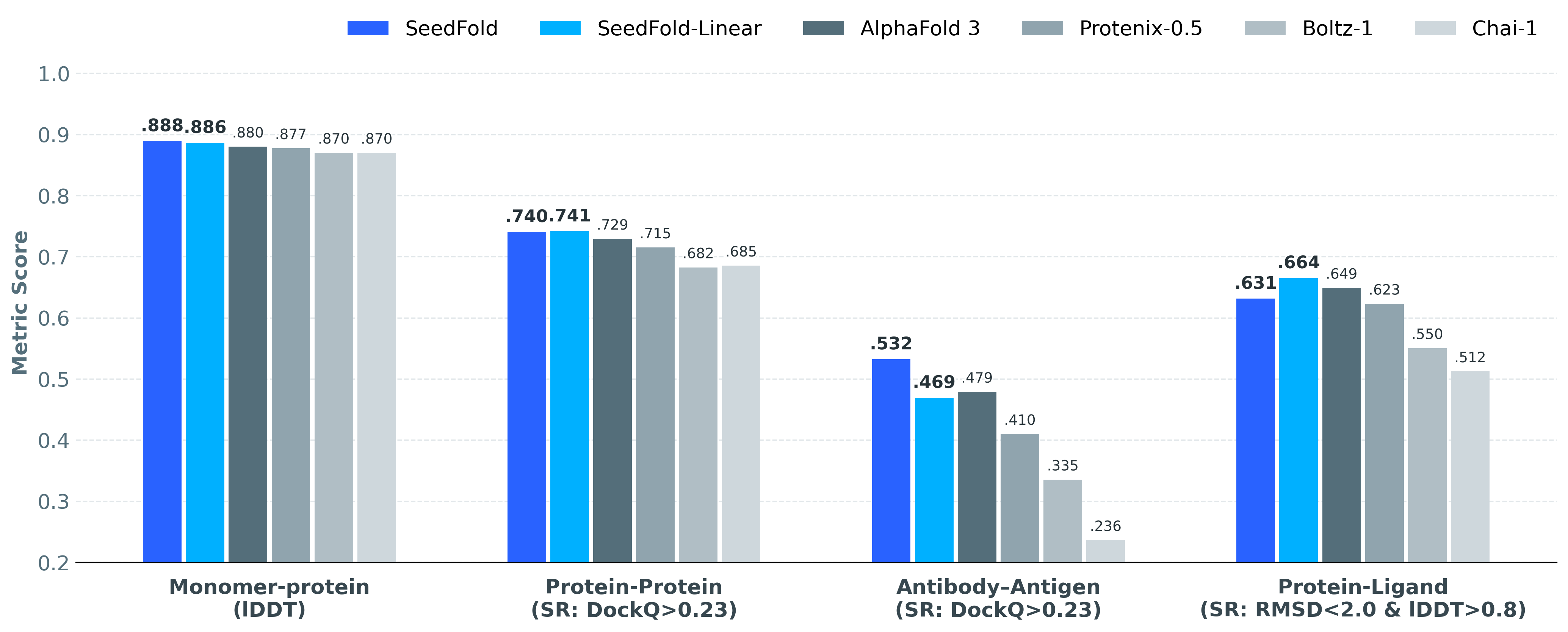
State-of-the-Art on FoldBench
Comprehensive evaluation across diverse biomolecular structure prediction tasks
| Task | SeedFold | SeedFold-Linear | AlphaFold 3 | Boltz-1 | Chai-1 | Protenix-0.5 |
|---|---|---|---|---|---|---|
| Monomer (lDDT) |
0.8889 | 0.8861 | 0.88 | 0.87 | 0.87 | 0.8773 |
| Protein-Protein (SR%: DockQ ≥ 0.23) |
74.03% | 74.14% | 72.93% | 68.25% | 68.53% | 71.50% |
| Ab-Ag (SR%: DockQ ≥ 0.23) |
53.21% | 46.91% | 47.90% | 33.54% | 23.64% | 41.00% |
| Protein-Ligand (SR%: lRMSD < 2Å and lDDT-PLI > 0.8) |
63.12% | 66.48% | 64.90% | 55.04% | 51.23% | 62.30% |
| Protein-RNA (SR%: DockQ ≥ 0.23) |
65.31% | 61.80% | 62.32% | 56.90% | 50.91% | 50.70% |
| Protein-DNA (SR%: DockQ ≥ 0.23) |
72.60% | 76.00% | 79.18% | 70.97% | 69.97% | 71.38% |
Method Overview
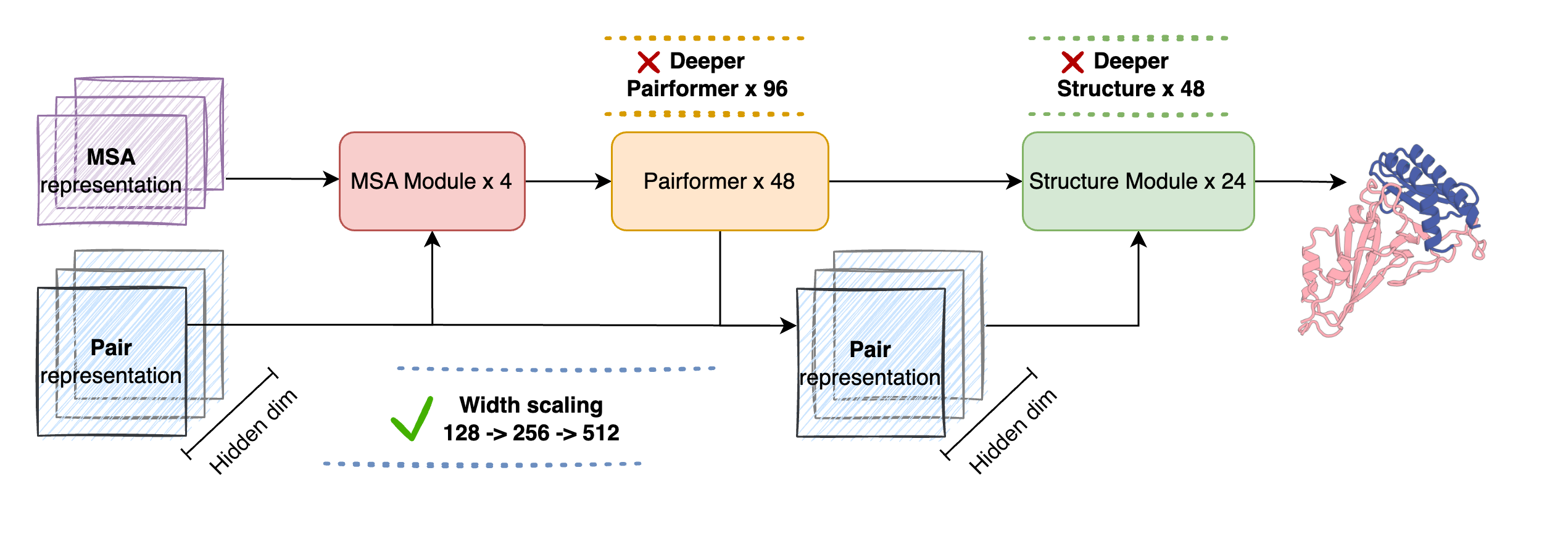
Model Architecture
SeedFold inherits the AlphaFold3 architecture with key modifications for scaling:
- MSA Module: Extracts evolutionary features from multiple sequence alignments
- Pairformer: Updates pair representations via triangular operations (scaled to 512 width)
- Structure Module: Diffusion-based all-atom structure generation
Width vs Depth Scaling
Our experiments demonstrate that width scaling of the Pairformer is the most effective strategy. The pair representation dimension is the critical bottleneck—increasing it directly enhances the model's capacity to encode complex pairwise interactions.
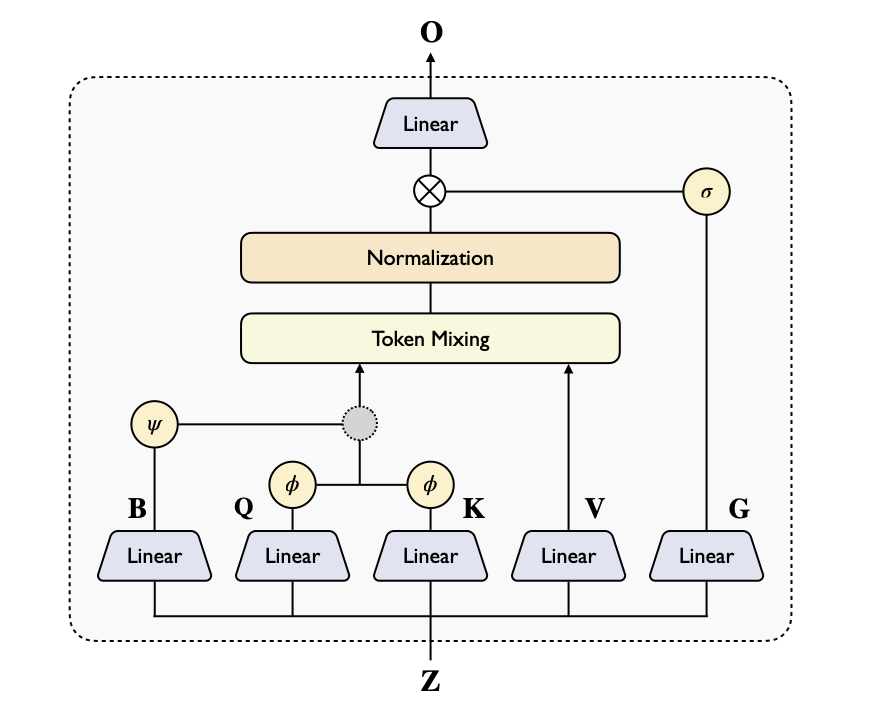
Linear Triangular Attention
We propose two variants of linear triangular attention to replace the computationally expensive vanilla triangular attention:
Additive Linear TriAtt
φ(Q)φ(K)ᵀ + ψ(B)
Inherits advantages of vanilla attention with well-established linear attention designs
Gated Linear TriAtt
φ(Q)φ(K)ᵀ ⊙ ψ(B)
Gating mechanism controls information flow; superior on DNA/RNA tasks
Contributors
Project Leads
*Equal contribution. Listing order is random.
Contributors
Technical Lead
ByteDance Seed
Citation
@misc{zhou2025seedfoldscalingbiomolecularstructure,
title={SeedFold: Scaling Biomolecular Structure Prediction},
author={Yi Zhou and Chan Lu and Yiming Ma and Wei Qu and Fei Ye and Kexin Zhang and Lan Wang and Minrui Gui and Quanquan Gu},
year={2025},
eprint={2512.24354},
archivePrefix={arXiv},
primaryClass={q-bio.BM},
url={https://arxiv.org/abs/2512.24354},
}SeedProteo Accurate De Novo All-Atom Design of Protein Binders
A diffusion-based model for de novo all-atom protein design. SeedProteo repurposes cutting-edge folding architecture into a powerful generative framework, achieving state-of-the-art performance in both unconditional generation and binder design.
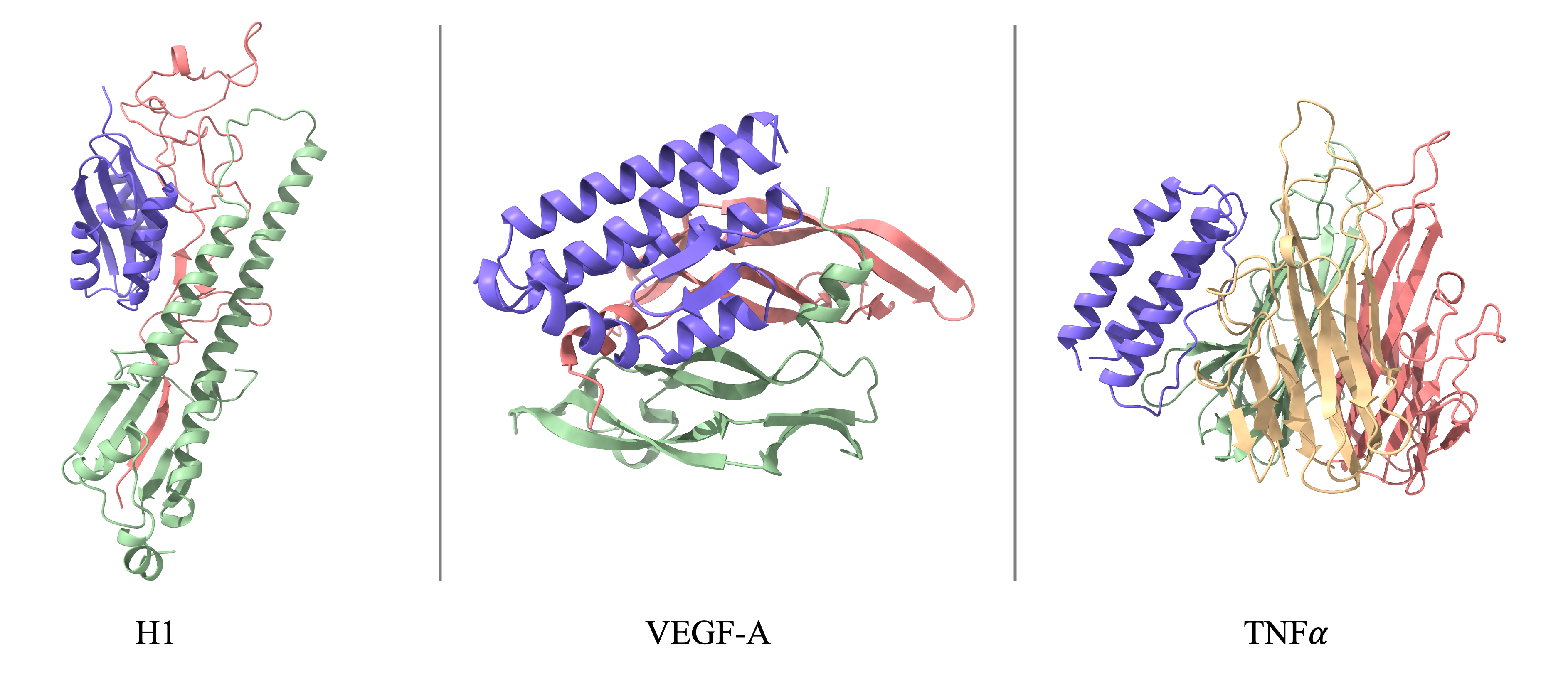
Binder Design Performance
SeedProteo outperforms open-source baselines across 10 benchmark targets
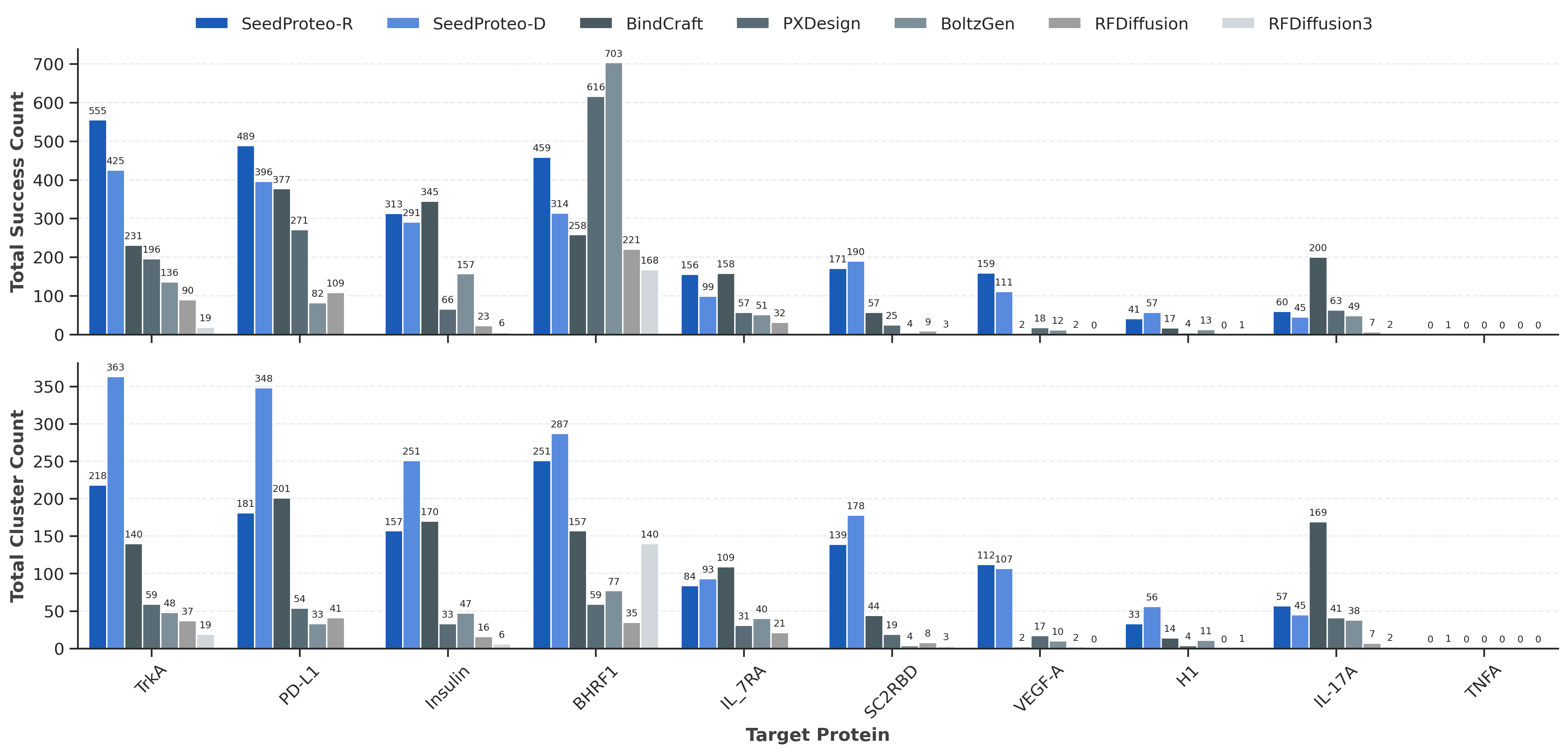
Comparison of binder design success and diversity. SeedProteo-R (Robust) and SeedProteo-D (Diverse) modes vs. baselines.
Structural Novelty in Binder Design
Lower novelty scores indicate more novel designs. Best performance per target in bold, second best underlined.
| Method | TrkA | PD-L1 | Insulin | BHRF1 | IL-7RA | SC2RBD | VEGF-A | H1 | IL-17A | TNFA |
|---|---|---|---|---|---|---|---|---|---|---|
| Ours | ||||||||||
| SeedProteo-D | 0.829 | 0.832 | 0.837 | 0.822 | 0.840 | 0.819 | 0.836 | 0.823 | 0.806 | 0.870 |
| SeedProteo-R | 0.905 | 0.913 | 0.911 | 0.872 | 0.917 | 0.858 | 0.901 | 0.890 | 0.855 | -- |
| Baselines | ||||||||||
| BindCraft | 0.849 | 0.856 | 0.864 | 0.847 | 0.861 | 0.863 | 0.850 | 0.830 | 0.818 | -- |
| PXDesign | 0.914 | 0.929 | 0.928 | 0.924 | 0.928 | 0.917 | 0.913 | 0.888 | 0.906 | -- |
| BoltzGen | 0.908 | 0.924 | 0.929 | 0.928 | 0.885 | 0.915 | 0.902 | 0.885 | 0.863 | -- |
| RFDiffusion | 0.932 | 0.934 | 0.927 | 0.946 | 0.916 | 0.912 | 0.940 | -- | 0.938 | -- |
| RFDiffusion3 | 0.808 | 0.834 | 0.876 | 0.845 | 0.840 | -- | -- | 0.930 | 0.800 | -- |
Technical Contributions
All-Atom Modeling
Directly designs proteins at the all-atom level using the atom14 schema, ensuring physically accurate structures.
- atom14 representation
- Full atomic detail
Self-Conditioning
Reuses denoised structures and predicted secondary structures to stabilize the sampling process.
- Design View integration
- Structure feedback
- Stable sampling
MRF Sequence Decoding
Uses Markov Random Field for energy minimization in sequence space, ensuring global consistency.
- Energy minimization
- Global consistency
- Optimal sequences
Methodology
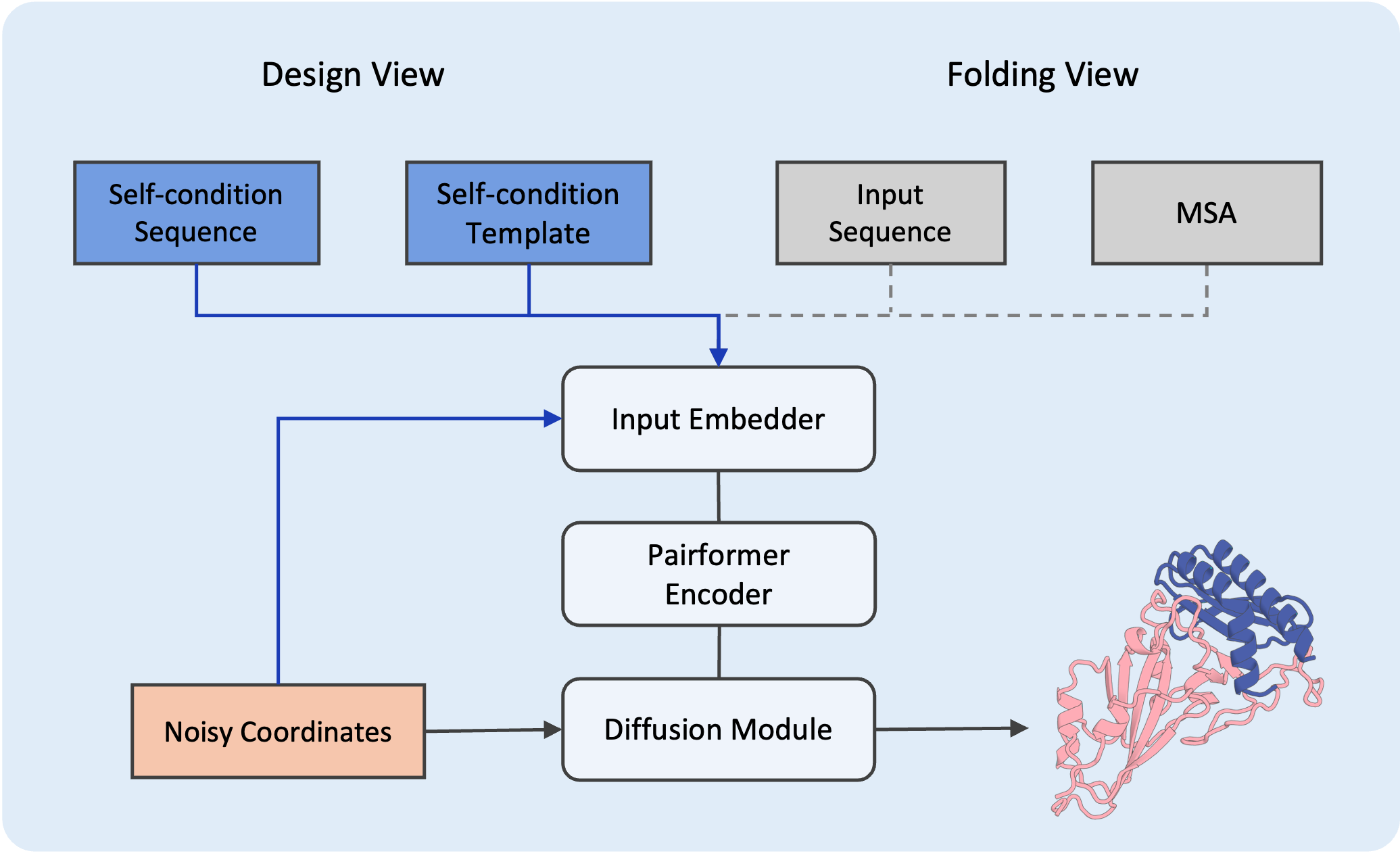
Design View Framework
SeedProteo mimics the architecture of AlphaFold3 and introduces a novel Design View that integrates self-conditioning features to guide the generative process.
- Left: Design View (Generative model)
- Right: Folding View (Input representation)
Unconditional Generation
Superior scalability, generating valid structures up to 1000 residues
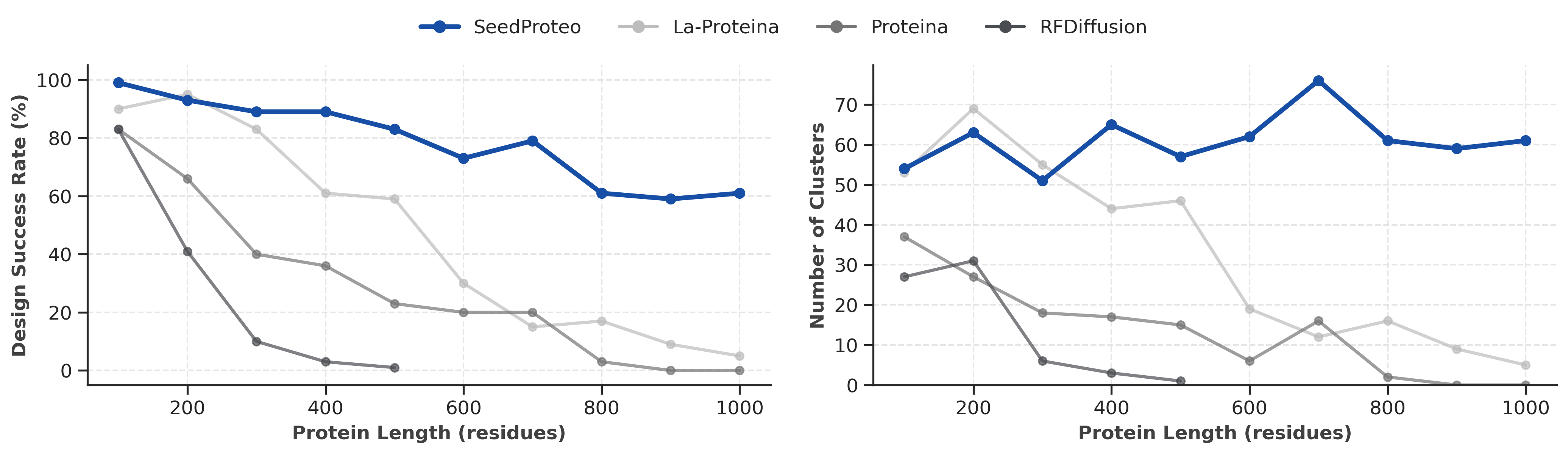
Unconditional monomer benchmark. Left: Design Success Rate vs Length. Right: Number of Unique Clusters vs Length.
Contributors
Project Lead
Contributors
†Work done during internship at ByteDance Seed
Technical Lead
ByteDance Seed
Citation
@misc{qu2025seedproteoaccuratenovoallatom,
title={SeedProteo: Accurate De Novo All-Atom Design of Protein Binders},
author={Wei Qu and Yiming Ma and Fei Ye and Chan Lu and Yi Zhou and Kexin Zhang and Lan Wang and Minrui Gui and Quanquan Gu},
year={2025},
eprint={2512.24192},
archivePrefix={arXiv},
primaryClass={q-bio.BM},
url={https://arxiv.org/abs/2512.24192},
}